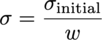ESPEI YAML input files¶
This page aims to completely describe the ESPEI input file in the YAML format. Possibly useful links are the YAML refcard and the (possibly less useful) Full YAML specification. These are all key value pairs in the format
key: value
They are nested for purely organizational purposes.
top_level_key:
key: value
As long as keys are nested under the correct heading, they have no required order. All of the possible keys are
system:
phase_models
datasets
tags
output:
verbosity
logfile
output_db
tracefile
probfile
generate_parameters:
excess_model
ref_state
ridge_alpha
mcmc:
iterations
prior
save_interval
cores
scheduler
input_db
restart_trace
chains_per_parameter
chain_std_deviation
deterministic
data_weights
The next sections describe each of the keys individually.
If a setting has a default of required it must be set explicitly.
system¶
The system key is intended to describe the specific system you are fitting, including the components, phases, and the data to fit to.
phase_models¶
| type: | string |
|---|---|
| default: | required |
The JSON file describing the CALPHAD models for each phase. See Phase Descriptions for an example of how to write this file.
datasets¶
| type: | string |
|---|---|
| default: | required |
The path to a directory containing JSON files of input datasets.
The file extension to each of the datasets must be named as .json, but they can otherwise be named freely.
For an examples of writing these input JSON files, see Making ESPEI datasets.
tags¶
| type: | dict |
|---|---|
| default: | required |
Mapping of keys to values to add to datasets with matching tags. These can be used to dynamically drive values in datasets without adjusting the datasets themselves. Useful for adjusting weights or other values in datasets in bulk. For an examples of using tags in input JSON files, see Tags.
output¶
verbosity¶
| type: | int |
|---|---|
| default: | 0 |
Controls the logging level. Most users will probably want to use Info or Trace.
Warning logs should almost never occur and this log level will be
relatively quiet. Debug is a fire hose of information, but may be useful in
fixing calculation errors or adjusting weights.
| Value | Log Level |
|---|---|
| 0 | Warning |
| 1 | Info |
| 2 | Trace |
| 3 | Debug |
logfile¶
| type: | string |
|---|---|
| default: | null |
Name of the file that the logs (controlled by verbosity) will be output to.
The default is None (in Python, null in JSON), meaning the logging will
be output to stdout and stderr.
output_db¶
| type: | string |
|---|---|
| default: | out.tdb |
The database to write out. Can be any file format that can be written by a pycalphad Database.
tracefile¶
| type: | string |
|---|---|
| default: | trace.npy |
Name of the file that the MCMC trace is written to.
The array has shape (number of chains, iterations, number of parameters).
The array is preallocated and padded with zeros, so if you selected to take 2000 MCMC iterations, but only got through 1500, the last 500 values would be all 0.
You must choose a unique file name.
An error will be raised if file specified by tracefile already exists.
probfile¶
| type: | string |
|---|---|
| default: | lnprob.npy |
Name of the file that the MCMC ln probabilities are written to.
The array has shape (number of chains, iterations).
The array is preallocated and padded with zeros, so if you selected to take 2000 MCMC iterations, but only got through 1500, the last 500 values would be all 0.
You must choose a unique file name.
An error will be raised if file specified by probfile already exists.
generate_parameters¶
The options in generate_parameters are used to control parameter selection and fitting to single phase data.
This should be used if you have input thermochemical data, such as heat capacities and mixing energies.
Generate parameters will use the Akaike information criterion to select model parameters and fit them, creating a database.
excess_model¶
| type: | string |
|---|---|
| default: | required |
| options: | linear |
Which type of model to use for excess mixing parameters. Currently only linear is supported.
The exponential model is planned, as well as support for custom models.
ref_state¶
| type: | string |
|---|---|
| default: | required |
| options: | SGTE91 | SR2016 |
The reference state to use for the pure elements and lattice stabilities. Currently only SGTE91 and SR2016 (for certain elements) is supported.
There are plans to extend to support custom reference states.
ridge_alpha¶
| type: | float |
|---|---|
| default: | 1.0e-100 |
Controls the ridge regression hyperparameter, $ alpha $, as given in the following equation for the ridge regression problem
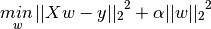
ridge_alpha should be a positive floating point number which scales the relative contribution of parameter magnitudes to the residuals.
If an exponential form is used, the floating point value must have a decimal place before the e,
that is 1e-4 is invalid while 1.e-4 is valid. More generally, the floating point must match the following
regular expression per the YAML 1.1 spec: [-+]?([0-9][0-9_]*)?\.[0-9.]*([eE][-+][0-9]+)?.
mcmc¶
The options in mcmc control how Markov Chain Monte Carlo is performed using the emcee package.
In order to run an MCMC fitting, you need to specify one and only one source of parameters somewhere in the input file.
The parameters can come from including a generate_parameters step, or by specifying the mcmc.input_db key with a file to load as pycalphad Database.
If you choose to use the parameters from a database, you can then further control settings based on whether it is the first MCMC run for a system (you are starting fresh) or whether you are continuing from a previous run (a ‘restart’).
iterations¶
| type: | int |
|---|---|
| default: | required |
Number of iterations to perform in emcee. Each iteration consists of accepting one step for each chain in the ensemble.
prior¶
| type: | list or dict |
|---|---|
| default: | {‘name’: ‘zero’} |
Either a single prior dictionary or a list of prior dictionaries corresponding to the number of parameters. See Specifying Priors for examples and details on writing priors.
save_interval¶
| type: | int |
|---|---|
| default: | 1 |
Controls the interval (in number of iterations) for saving the MCMC chain and probability files. By default, new files will be written out every iteration. For large files (many mcmc iterations and chains per parameter), these might become expensive to write out to disk.
cores¶
| type: | int |
|---|---|
| min: | 1 |
How many cores from available cores to use during parallelization with dask or emcee. If the chosen number of cores is larger than available, then this value is ignored and espei defaults to using the number available.
Cores does not take affect for MPIPool scheduler option. MPIPool requires the number of processors be set directly with MPI.
scheduler¶
| type: | string |
|---|---|
| default: | dask |
| options: | dask | None | JSON file |
Which scheduler to use for parallelization. You can choose from either dask, None, or pass the path to a JSON scheduler file created by dask-distributed.
Choosing dask allows for the choice of cores used through the cores key.
Choosing None will result in no parallel scheduler being used. This is useful for debugging.
Passing the path to a JSON scheduler file will use the resources set up by the scheduler.
JSON file schedulers are most useful because schedulers can be started on MPI clusters using dask-mpi command.
See Advanced Schedulers for more information.
input_db¶
| type: | string |
|---|---|
| default: | null |
A file path that can be read as a pycalphad Database. The parameters to fit will be taken from this database.
For a parameter to be fit, it must be a symbol where the name starts with VV, e.g. VV0001.
For a TDB formatted database, this means that the free parameters must be functions of a single value that are used in your parameters.
For example, the following is a valid symbol to fit:
FUNCTION VV0000 298.15 10000; 6000 N !
restart_trace¶
| type: | string |
|---|---|
| default: | null |
If you have run a previous MCMC calculation, then you will have a trace file that describes the position and history of all of the chains from the run.
You can use these chains to start the emcee run and pick up from where you left off in the MCMC run by passing the trace file (e.g. chain.npy) to this key.
If you are restarting from a previous calculation, you must also specify the same database file (with input_db) as you used to run that calculation.
chains_per_parameter¶
| type: | int |
|---|---|
| default: | 2 |
This controls the number of chains to run in the MCMC calculation as an integer multiple of the number of parameters.
This parameter can only be used when initializing the first MCMC run. If you are restarting a calculation, the number of chains per parameter is fixed by the number you chose previously.
Ensemble samplers require at least 2*p chains for p fitting parameters to be able to make proposals.
If chains_per_parameter = 2, then the number of chains if there are 10 parameters to fit is 20.
The value of chains_per_parameter must be an EVEN integer.
chain_std_deviation¶
| type: | float |
|---|---|
| default: | 0.1 |
The standard deviation to use when initializing chains in a Gaussian distribution from a set of parameters as a fraction of the parameter.
A value of 0.1 means that for parameters with values (-1.5, 2000, 50000) the chains will be initialized using those values as the mean and (0.15, 200, 5000) as standard deviations for each parameter, respectively.
This parameter can only be used when initializing the first MCMC run. If you are restarting a calculation, the standard deviation for your chains are fixed by the value you chose previously.
You may technically set this to any positive value, you would like. Be warned that too small of a standard deviation may cause convergence to a local minimum in parameter space and slow convergence, while a standard deviation that is too large may cause convergence to meaningless thermodynamic descriptions.
deterministic¶
| type: | bool |
|---|---|
| default: | True |
Toggles whether ESPEI runs are deterministic. If this is True, running
ESPEI with the same Database and initial settings (either the same
chains_per_parameter and chain_std_deviation or the same
restart_trace) will result in exactly the same results.
Starting two runs with the same TDB or with parameter generation (which is deterministic) will result in the chains being at exactly the same position after 100 iterations. If these are both restarted after 100 iterations for another 50 iterations, then the final chain after 150 iterations will be the same.
It is important to note that this is only explictly True when starting at the same point. If Run 1 and Run 2 are started with the same initial parameters and Run 1 proceeds 50 iterations while Run 2 proceeds 100 iterations, restarting Run 1 for 100 iterations and Run 2 for 50 iterations (so they are both at 150 total iterations) will NOT give the same result.
data_weights¶
| type: | dict |
|---|---|
| default: | {‘ZPF’: 1.0, ‘ACR’: 1.0, ‘HM’: 1.0, ‘SM’: 1.0, ‘CPM’: 1.0} |
Each type of data can be weighted: zero phase fraction (ZPF), activity
(ACR) and the different types of thermochemical error. These weights are
used to modify the initial standard deviation of each data type by